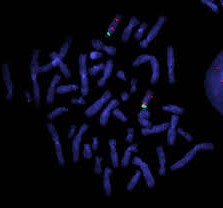
Los síndromes de Prader Willi (SPW) y de Angelman (SA) son afecciones neurogenéticas con múltiples manifestaciones fenotípicas, que «mapean» en la misma región del cromosoma 15. El SPW tiene una incidencia estimada en 1/10.000 a 1/15.000 nacidos vivos, mientras que el SA se presenta con una frecuencia de 1/12.000 a 1/20.000 nacidos vivos.
En ambos cuadros el diagnóstico clínico se fundamenta en características fenotípicas variables e inespecíficas, especialmente durante el primer año de vida. En el caso del SPW, el diagnóstico se sospecha en todo recién nacido con hipotonía y dismorfias craneofaciales menores.
En los casos de SA, habitualmente el diagnóstico es más tardío, cercano a la edad preescolar. En niños con afectación seria se presentan con retraso severo del desarrollo, microcefalia, compromiso extrapiramidal, macrostomía, prognatismo, risa espasmódica, ausencia del lenguaje y alteraciones electroencefalográficas, con o sin convulsiones. En nuestro instituto el diagnóstico de ambos síndromes se realiza mediante PCR utilizando partidores específicos para las secuencias metiladas y no metiladas del gen SNRPN.
Dra. Vanesa Beretta
(Dpto. Biología Molecular)